Exercise-induced Vascular Adaptations Modulating Angiotensin II Responses
Authors: Agnaldo Bruno Chies1*, Luis Gabriel Navar2
1Laboratory of Pharmacology, Marilia Medical School, Marilia, Sao Paulo, Brazil
2Department of Physiology and Hypertension and Renal Center of Excellence, Tulane University School of Medicine, New Orleans, LA, USA.
*Correspondence to: Agnaldo Bruno Chies, Laboratory of Pharmacology, Marilia Medical School, Marilia, Sao Paulo, Brazil; E-mail: agnaldo.chies.abc@gmail.com
Received: August 08, 2021; Revision: November 19, 2021; Published: November 27, 2021
Citation: Chies AB and Navar lG (2021) Exercise-induced Vascular Adaptations Modulating Angiotensin II Responses. 21st Century Cardiol, Volume 1 (2): 108
Abstract
The renin-angiotensin system (RAS) acts in different physiological processes to maintain renal perfusion and blood pressure. In this context, it regulates the vascular tonus through the actions of angiotensin II (Ang II) on AT1 receptors (AT1R), which evoke vasoconstriction. The Ang II-induced vasoconstriction, however, must be modulated by endothelial mechanisms to avoid excess reductions in blood flow and cardiovascular injury. This endothelial modulation of Ang II responses, in turn, may be enhanced by regular physical exercise. This phenomenon is well characterized in the arterial bed but less well characterized in the venous bed. Our recent studies have addressed this knowledge gap and demonstrated the complex and variable modulatory influences on different venous beds. These results provide a deeper understanding of the influence of exercise on the adaptations occurring in the venous bed that provide therapeutic benefits to the entire cardiovascular system.
Keywords
Angiotensin II; Endothelium; Exercise; Vasoconstriction; Vein
1. Renin-angiotensin system: concepts and functions
The renin-angiotensin system (RAS) is an endocrine system involved in many functions throughout the body including the maintenance of renal perfusion and arterial blood pressure. This system is activated when the juxtaglomelular apparatus secretes renin in response to a reduction in the concentration of sodium chloride at the macula densa, adrenergic stimuli, or by barosensor mechanisms present in the afferent arterioles [1,2]. Once released, this enzyme converts the liver-produced angiotensinogen to angiotensin I, which in turn, is converted to angiotensin II (Ang II) by the angiotensin-converting enzyme-1 (ACE1), which is on most endothelial cells throughout the body but is predominantly anchored in the endothelium of pulmonary vessels. Once formed, Ang II promotes many important physiological effects acting mainly on AT1 receptors (AT1R) in various tissues. Among the main Ang II-induced actions on the kidneys is the regulation of glomerular filtration rate, renal perfusion, tubular sodium reabsorption, directly or through the release of aldosterone from the adrenal gland cortex, the facilitation of adrenergic neurotransmission, and/or the vasoconstriction that contributes to the maintenance of global vascular tone and peripheral vascular resistance [3].
The concept of the RAS as a purely endocrine system has been modified in recent years because of evidence that RAS components such as Ang II and other pharmacologically active peptides are produced locally to act in a paracrine or autocrine manner [3]. In this context, it was observed that intrarenal Ang II concentrations and the blood pressure are elevated during the initial period of 2-Kidney-1-Clip (2K1C) hypertension in rats and remains elevated despite the restoration of the concentrations of plasma Ang II and plasma renin activity to the normal range by 25 days after clipping [4]. Evidence of
Ang II synthesis in the nephrons as well as the production of angiotensinogen by proximal tubule cells and of renin by the main cells of connecting tubules [5,6] also complement the classical view of RAS.
One unquestionable role of the RAS is the maintenance of circulatory homeostasis and glomerular function, especially in the face of reductions in blood volume. For reasons not yet completely understood, however, this system may become inappropriately activated and exert pathophysiological actions, thus contributing to the development of cardiovascular injuries such as the direct participation of the RAS in the pathophysiology of arterial hypertension, cardiac hypertrophy, and heart failure. Through the inappropriate activation of AT1R, Ang II promotes hypertrophy, fibrosis, atherosclerosis, inflammation, and other vascular and interstitial injuries. The action of Ang II on AT1R also stimulates superoxide anion production, thereby resulting in oxidative stress [7]. It is precisely in the inhibition of the ACE1 - Ang II - AT1R axis, through ACE inhibitors or AT1R antagonists that a large assortment of pharmacological treatments for cardiovascular and inflammatory diseases is based on targeting this system. It is noteworthy, however, that the actions of the RAS are intrinsically counterbalanced by the axis ACE2 ? Ang (1-7) ? Mas receptor, which most often exerts actions opposite to the axis ACE1 - Ang II ? AT1R [3]. Possibly, many cardiovascular diseases result from the disruption of this delicate balance between axes. Agonist drugs of Mas receptors may become an important therapy for treating cardiovascular diseases.
While Ang II exerts an important role in the control of vascular tone, the actions of Ang II are modulated by endothelium-derived vasodilator (mainly) and vasoconstrictor factors. This tight local control provides a counterbalancing action to prevent an uncontrolled rise in peripheral vascular resistance that could drive systemic blood pressure to dangerous hypertensive levels and reduce blood flow to the extremities of the body. Ang II acts on AT1R on vascular smooth muscle, promoting vasoconstriction by activating the phosphokinase C (PKC) ? inositol triphosphates (IP3)/diacylglycerol (DAG) pathway, which culminates in intracellular calcium elevation [3]. This vasoconstrictor action resulting from the activation of AT1R, on the other hand, is counterbalanced by the action of Ang II on AT2 receptors (AT2R) present in the vascular endothelium, which culminates in the release of relaxing factors such as nitric oxide (NO) [3,8]. These substances can also be released through pathways that do not involve AT2R such as increased shear stress and activation of Mas receptors by ACE2 derived Ang [1-7]. In this manner, the vasoconstrictor influence exerted by Ang II on the control of vascular tone is the result of the vasoconstrictor action mediated by AT1R but modulated by predominantly vasodilator substances that are released by the endothelium.
2. Influence of exercise on vascular responses to angiotensin II
The local modulation of Ang II vasoconstrictor actions is essential to provide a balance of forces that determine peripheral vascular resistance. To preserve cardiovascular health, several therapeutic strategies aimed at improving the function of these modulating mechanisms have been studied. These strategies, in general, increase the production of local modulating substances, such as replacement of estrogenic hormones, treatment with statins, as well as prevention of degradation of vasodilator factors through the use of antioxidants or by restricting the use of non-steroidal anti-inflammatory drugs [9]. RAS blockers are the major strategy to reduce the Ang II influence since they inhibit the formation of Ang II or reduce the AT1R activation or expression of NADPH oxidases and, consequently, the production of the superoxide anion, substances that reduce the bioavailability of nitric oxide (NO) [7].
In this context, regular physical exercise has a prominent place as an important strategy for the prevention and treatment of cardiovascular diseases, often in association with pharmacological therapies. The benefits of physical exercise occur in different organs and systems and are not restricted to the cardiovascular system. The effects of physical exercise on the mechanisms that regulate vascular tone are broad and well documented. Physical exercise either increases the biosynthesis of endothelium-derived substances such as NO or prevents their degradation, thereby improving the local modulation of the actions of mediators involved in the regulation of vascular tone. Possibly, the repeated shear stress elevations that occur as a result of circulatory changes promoted by regular physical exercise increase the expression/activity of enzymes that produce these modulating substances, especially nitric oxide synthases (NOS) and cyclooxygenases (COX). In parallel, the exercise-related shear stress is also capable of expressing enzymes that increase the degradation of reactive oxygen species, which contribute to the reduction of oxidative stress and increase the bioavailability of NO. Notably, regular physical exercise leads to improvements in the endothelial mechanisms that modulate the responses to Ang II both in large-conductance arteries [8] and in the microcirculation [10]. These exercise-induced endothelial adaptations may influence the effects of other vasoconstrictors (catecholamines, endothelin-1, and others) which, in parallel to Ang II, determine the overall vasomotor tone. These circulatory adaptations are part of a set of physiological adjustments that allow the body to more efficiently direct blood flow to tissues where metabolic demand is increased by exercise [11]. In essence, cardiovascular benefits occur as the body adapts to support the stress imposed by exercise.
Exercise-induced circulatory redistribution, however, is not restricted to the arterial microvasculature. To meet the metabolic needs imposed by exercise, large blood volumes are displaced from the main capacitance compartments as well as from the vascular beds of inactive regions toward tissues most metabolically involved in the physical exercise [12]. In this context, the importance of the venous bed cannot be ignored since about 60-80% of the blood in mammals is located in the venous compartment and a significant part of this volume is shifted to the heart and skeletal muscles as a consequence of exercise-induced circulatory redistribution [13]. As in arterial beds, adaptations in local mechanisms that modulate the actions of vasoconstrictor mediators can also occur in venous beds. Despite this, most studies that aim to better understand the effects of physical exercise on these endothelium-related modulatory mechanisms have been focused primarily or exclusively on the arterial vasculature, and very little is known about the venous bed. Completing this important knowledge gap will not only allow a deeper understanding of exercise physiology but may provide evidence that adaptations occurring in the venous bed bring therapeutic benefits of exercise to the entire cardiovascular system.
Moved by this challenge, we have focused efforts in recent years to investigate the changes associated with physical exercise in the responses of isolated rat veins to Ang II, seeking to understand the participation of NO, prostanoids, and endothelin-1 in this process. We initially observed that repeated physical exercise adapts the rat portal vein to vigorously respond to Ang II. This vascular adaptation is not agonist-specific. It influences both Ang II and phenylephrine-induced responses but is territory-specific because it was not observed in the vena cava. In addition, it appears to involve NO and prostanoids, which are prevalent during rest, and endothelin-1, which prevails after exercise [14,15]. These adaptations allow the blood present in the distal portion of the splanchnic territory and the liver to be displaced to the heart and skeletal muscle more efficiently.
Further studies were focused on venous beds that exert distinct physiological roles from the portal vein. This is related to an interesting debate regarding the role of active venoconstriction in the exercise-induced increase of the end-diastolic volume. Some authors argue that the active venoconstriction contributes to the blood shifting toward the heart since it increases the stressed volume in the venous compartment [16]. Others, instead, point out that active venoconstriction could even impair the venous return since it may cause a proportionally much larger alteration in venous resistance to flow [17]. Thus, assuming that venoconstriction can impair venous return depending on the bed, we decided to investigate the modulating mechanisms of Ang II venoconstrictor responses in the femoral vein. We chose to study this vessel assuming that uncontrolled vasoconstriction there would possibly impair the return of blood from the animals' hind legs. The obtained results suggest that, unlike what was observed in the portal vein, both acute or repeated exercise reinforce Ang II-mediated suppression in the femoral vein. We observed that vasodilator prostanoids and other vasodilator mechanisms triggered by the action of endothelin-1 on ETB receptors, act as a backup to NO in animals exposed to exercise, and thus maintain control of the Ang II responses in femoral veins (Figure 1) [18]. Interestingly, a similar exercise-induced adaptation was observed in femoral veins from rats with 2K1C mediated hypertension. Possibly, in these 2K1C animals who?s RAS is even more active, these adaptations are essential to ensure an adequate venous return [19].
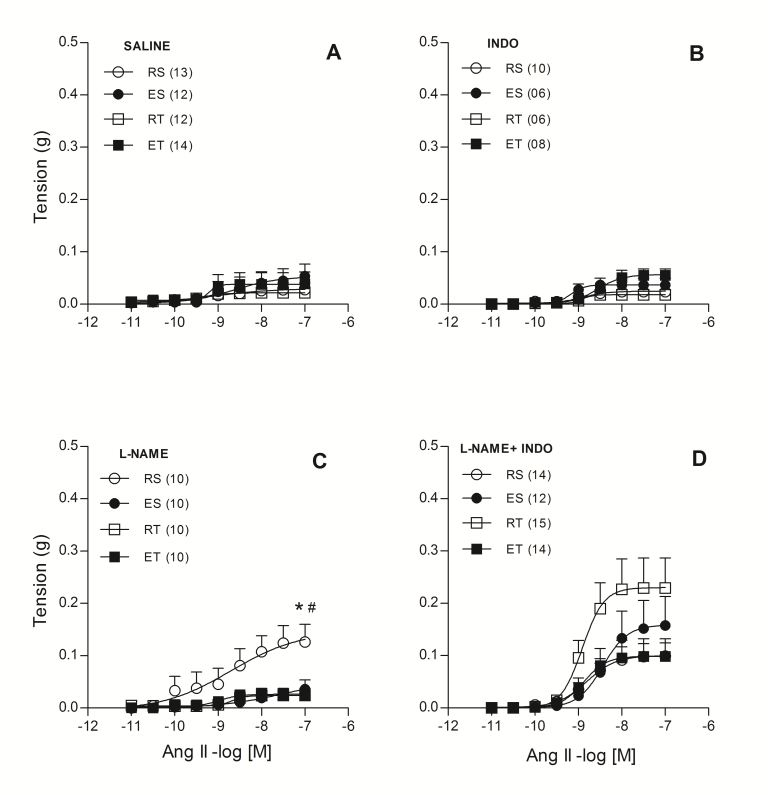
Figure 1: Ang II concentration-response curves determined in circular preparations of the femoral vein taken from resting-sedentary (RS), exercised-sedentary (ES), resting-trained (RT) and exercised-trained (ET) rats, treated with saline (A), 10-5 mol/L indomethacin (INDO; B), 10-4 mol/L L-NAME (C) or 10-4 mol/L L-NAME and 10-5 mol/L INDO (D). Points represent the mean ? SEM, and the number of independent determinations is in parentheses. * indicates a significant difference (P<0.01) in relation to the ES animals; # indicates a significant difference (P<0.01) in relation to the RT animals, respectively (two-way ANOVA followed by Bonferroni?s post-test) [18].
The effects of exercise on the Ang II responses have also been investigated in mesenteric veins that drain blood from the splanchnic region. Curiously, the role of active vasoconstriction in blood return is more consensual in this vascular territory. In this isolated vein, the exercise training mobilizes endothelin-1 to reinforce the Ang II-induced responses. On the other hand, vasodilator prostanoids are mobilized to act in parallel with NO to counterbalance the Ang II responses that have been potentiated by endothelin-1 in the mesenteric veins of these trained animals [20]. These show the importance of vasoconstriction in the blood displacement from the splanchnic territory but also reinforce the importance of adequate control of Ang II responses for proper circulatory functioning.
Conclusion
In conclusion, it has already been established that physical exercise modulates vascular responses to Ang II as well as to other vasoconstrictors, thereby ensuring adequate vascular resistance and venous return. These adaptations make the exercise-induced circulatory redistribution more efficient and, thus, adapt the organism to the stress imposed by future exercise exposition. Furthermore, these adaptations have a therapeutic role, reducing the incidence of cardiovascular diseases and/or contributing to their resolution. These exercise-induced adaptations are well characterized in the arterial bed but less well characterized in the venous bed. Our recent studies have addressed the knowledge gap and demonstrated the complex and variable modulatory influences on different venous beds. These results provide a deeper understanding of the influence of exercise on the adaptations occurring in the venous bed that provides therapeutic benefits to the entire cardiovascular system (Figure 2).
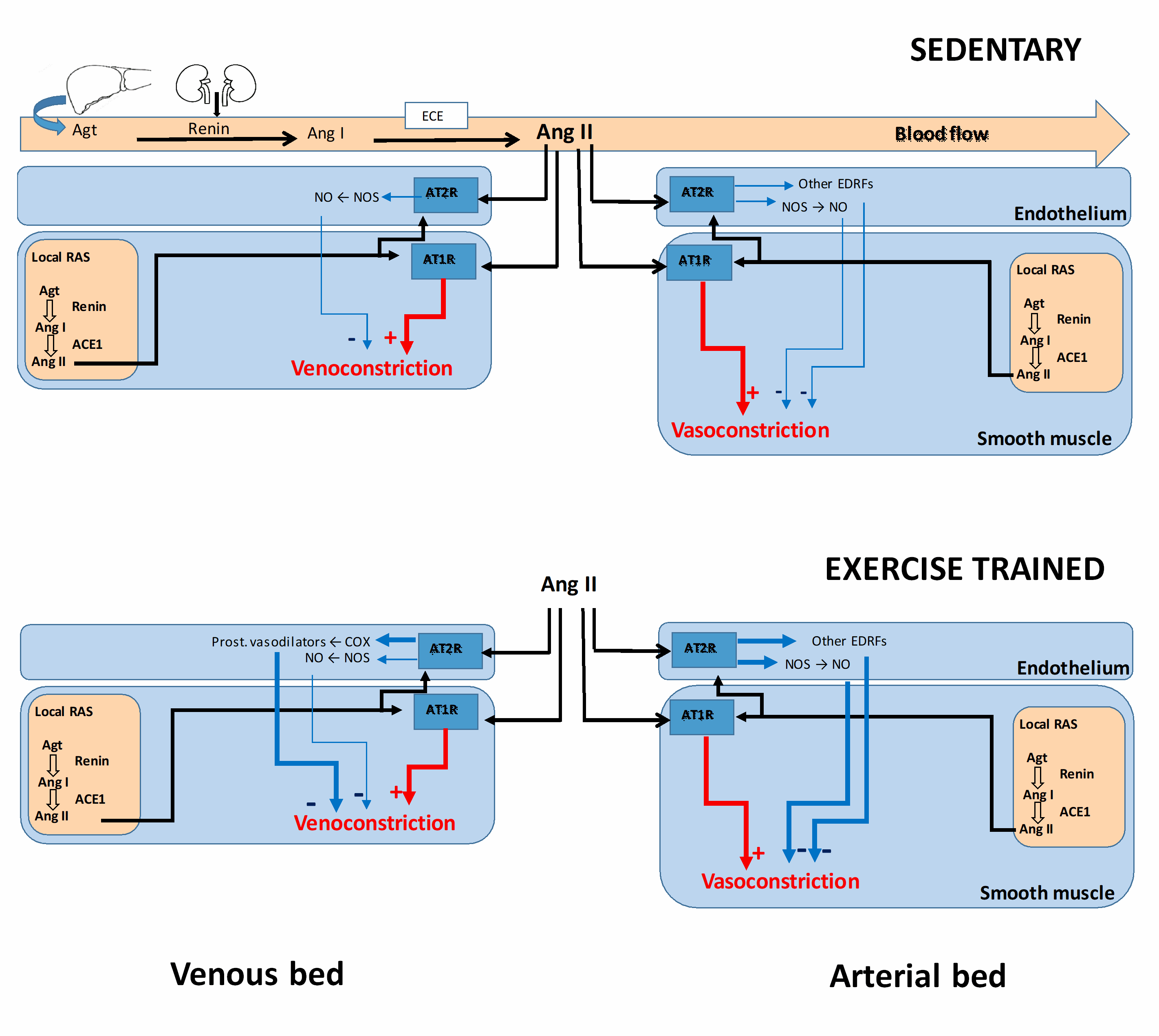
Figure 2: Modulations of angiotensin II-induced vasoconstriction in both veins and arteries. The Renin-Angiotensin System (RAS) may act in an endocrine (circulating RAS) or paracrine/autocrine fashion. In both cases, angiotensinogen (Agt) is converted in angiotensin I (Ang I) by renin that, which in turn, is converted in angiotensin II (Ang II) by angiotensin converting enzyme-1 (ACE). Several vasodilator endothelial mechanism modulate the angiotensin II-induced vasoconstrictor, such as nitric oxide (NO) formed by the enzymes nitric oxide synthases (NOS), vasodilator prostanoids formed by cyclooxygenase enzymes (COX), as well as other endothelium-derived relaxing factors (EDRF). Under training, these vasodilator endothelial mechanisms may be improved to to keep controlled the Ang II responses.
References
1. Sparks MA, Crowley SD, Gurley SB, et al. (2014) Classical renin-angiotensin system in kidney physiology. Compr Physiol 4: 1201-1228. https://dx.doi.org/10.1002/cphy.c130040
2. Kopp UC (2011) Integrated systems physiology: From molecule to function to disease. Neural control of renal function. San Rafael (CA): Morgan & Claypool Life Sciences.
3. Karnik SS, Unal H, Kemp JR, et al. (2015) International union of basic and clinical pharmacology. XCIX. Angiotensin receptors: Interpreters of pathophysiological angiotensinergic stimuli [corrected]. Pharmacol Rev 67: 754-819. https://doi.org/10.1124/pr.114.010454
4. Guan S, Fox J, Mitchell KD, et al. (1992) Angiotensin and angiotensin converting enzyme tissue levels in two-kidney, one clip hypertensive rats. Hypertension 20: 763-767. https://doi.org/10.1161/01.hyp.20.6.763
5. Ichihara A, Kobori H, Nishiyama A, et al. (2004) Renal renin-angiotensin system. Contrib Nephrol 143: 117-130. https://doi.org/10.1159/000078716
6. Navar LG, Kobori H, Prieto MC, et al. (2011) Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355-362. https://dx.doi.org/10.1161/HYPERTENSIONAHA.110.163519
7. Forrester SJ, Booz GW, Sigmund CD, et al. (2018) Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev 98: 1627-1738. https://doi.org/10.1152/physrev.00038.2017
8. Oliveira PR, Oliveira PB, Rossignoli PS, et al. (2020) Exercise training attenuates angiotensin II-induced vasoconstriction in the aorta of normotensive but not hypertensive rats. Exp Physiol 105: 732-742. https://doi.org/10.1113/ep088139
9. Cau SB, Carneiro FS, Tostes RC (2012) Differential modulation of nitric oxide synthases in aging: therapeutic opportunities. Front Physiol 3: 218. https://doi.org/10.3389/fphys.2012.00218
10. Park Y, Prisby RD, Behnke BJ, et al. (1985) Effects of aging, TNF-alpha, and exercise training on angiotensin II-induced vasoconstriction of rat skeletal muscle arterioles. J Appl Physiol (1985) 113: 1091-1100. https://doi.org/10.1152/japplphysiol.00292.2012
11. Armstrong RB, Delp MD, Goljan EF, et al. (1987) Distribution of blood flow in muscles of miniature swine during exercise. J Appl Physiol (1985) 62: 1285-1298. https://doi.org/10.1152/jappl.1987.62.3.1285
12. Musch TI, Friedman DB, Pitetti KH, et al. (1987) Regional distribution of blood flow of dogs during graded dynamic exercise. J Appl Physiol (1985) 63: 2269-2277. https://doi.org/10.1152/jappl.1987.63.6.2269
13. Rothe CF (1983) Venous system: physiology of the capacitance vessels. In: Sherpherd JT, Abboud FM, editors. Handbook of physiology: The cardiovascular system. Washington (DC). Wiley-Blackwell. 397-452.
14. Chies AB, de Souza Rossignoli P (2009) Exercise increases the phenylephrine effects in isolated portal vein of trained rats. Vascul Pharmacol 51: 125-132. https://doi.org/10.1016/j.vph.2009.05.003
15. Chies AB, de Souza Rossignoli P, Daniel EF (2010) Exercise increases the angiotensin II effects in isolated portal vein of trained rats. Peptides 31: 883-888. https://doi.org/10.1016/j.peptides.2010.02.011
16. Rothe CF (2006) Point: active venoconstriction is/is not important in maintaining or raising end-diastolic volume and stroke volume during exercise and orthostasis. J Appl Physiol (1985) 101: 1262-1264; discussion 1265-1266, 12770. https://doi.org/10.1152/japplphysiol.00561.2006
17. Hainsworth R, Drinkhill MJ (2006) Counterpoint: active venoconstriction is not important in maintaining or raising end-diastolic volume and stroke volume during exercise and orthostasis. J Appl Physiol (1985) 101: 1264-1265; discussion 1265-1266, 12770.
18. Chies AB, Rossignoli Pde S, Baptista Rde F, de Labio RW, et al. (2013) Exercise reduces angiotensin II responses in rat femoral veins. Peptides 44: 47-54.
19. Chies AB, Spadella MA, de Oliveira PR, et al. (2021) Exercise-induced modulation of Angiotensin II responses in femoral veins from 2-Kidney-1-Clip hypertensive rats. Front Physiol 12: 620438.
20. Chies AB, de Oliveira PB, Rossignoli PS, et al. (2017) Prostanoids counterbalance the synergism between endothelin-1 and angiotensin II in mesenteric veins of trained rats. Peptides 88: 67-73.