Biventricular Thrombi in New Onset Dilates Cardiomyopathy : an Unusual Presentation of Behcet Disease
Authors: Imane Katif*
178 riad salam, Marrakech, Morocco; katif.imane@gmail.com
*Corresponding author: Imane Katif, 178 riad salam, Marrakech, Morocco; katif.imane@gmail.com
Received: 08 April 2023; Accepted: 25 April 2023; Published: 28 April 2023
Citation: Katif I, (2023) Biventricular Thrombi in New Onset Dilates Cardiomyopathy: An Unusual Presentation of Behcet Disease 21st Century Cardiol, Volume 3 (2): 132
Abstract
Behcet’s disease is a systemic vasculitis of the vessels for all calibers, touching arterial and venous territories. The causes of disease are unknow. BD reaches young age subjects from 10 to 45 years and affects both men and women. BD is ubiquitous but more frequent in patients from Mediterranean basin, the middle East and Asia. The diagnosis of BD is essentially clinical. The diagnostic criteria make it possible to carry the diagnosis with good sensitivity and specifity. BD evolves by recurrent inflammatory attack. BD can affect all of the organs; cardiacs manifestations are dominated by intracardiac thrombosis, the damage of three tunics, coronaryarteritis with or without myocardial infarction, coronaries aneurysms and endomyocardial fibrosis. The vascular manifestations are dominated by arterial or venous thrombosis. The presence of dilated cardiomyopathy with reduced left ventricular ejection fraction is rare. It can be explained by ischemic or inflammatory origin by cytokines. We report a case of young woman aged of 33 years to the history of 3 episodes of bipolar aphtae which presented dilated cardiomyopathy with reduced left ventricular function, biventricular thrombosis, bilateral distal pulmonary embolism with pulmonary infarction.
Keywords:
Dilated cardiomyopathy; Behcet disease; Thrombus; Left ventricular dysfonction
Introduction
The BD is a rare systemic vasculitis of unknown cause which is characterized by recurrent inflammatory attacks affecting many organs [1]. Pathophysiology hallmark is an occlusive vasculitis local and systemic. BD is observed worldwide but more frequent in countries located on the old silk road especially in the middle East, Japan, Turkey. In Africa the cases are rare essentially found in the Maghreb [1-3]. BD often touch young patients aged between 30 and 40 years, it is exceptional before puberty or after age of 50 year. It usually affects young subjects between 30 and 40 years old, is rare before puberty and after 50 years [1-4]. It is associated with significant morbidity and mortality particularly in men when it occurs at a very young age [1-4]. The main manifestations are mucocutaneous, ocular, joint, neurological, vascular (venous and arterial) and cardiac involvement [1,3,4]. Cardiac involvement is dominated by intracardiac thrombosis, tunica involvement, coronary arteritis and aneurysms, and endomyocardial fibrosis [1,3,4]. The diagnosis is clinical. Treatment is symptomatic with anti-inflammatory and/or immunosuppressive drugs [1,5].
Patient and observation
The patient was 33 years old, with no particular cardiovascular risk factors apart from gestational diabetes, with no known cardiac disease, 3 episodes of bipolar aphthosis per year, on the oestroprogestogenic pill for 1 month, and no history of miscarriage (last child aged 17 months). For the past 8 months, she has had NYHA stage II dyspnoea, aggravated 15 days before her admission to the emergency room by rest dyspnoea with productive cough, moderate haemoptysis in a context of fever with poor general condition. She was hemodynamically stable with a blood pressure of 110/70 mmHg, polypnoea at 32 cycles/min, tachycardia at 110 bpm. The examination revealed bilateral crackles, signs of right heart failure with oedema of the lower limbs, and turgid jugular veins. The electrocardiogram shows sinus tachycardia, left anterior hemiblock. Previously, the patient had presented to the emergency department and the diagnosis of bilateral distal pulmonary embolism with an area of infarction had been made after a chest CT angiogram (Figure 1) with diffuse ground glass images; the diagnosis of Covid-19 infection was ruled out after negative PCR and serology. Biologically, there was a biological inflammatory syndrome with CRP 88 mg/l, leukocytes 9.8 giga/l, platelets 289 giga/l, moderate impairment of renal function with a clearance of 42 ml/min. The pathergy test was positive. Immunological assessment and Serological test were negative. The BK sputum to eliminate atypical tuberculosis came back negative.
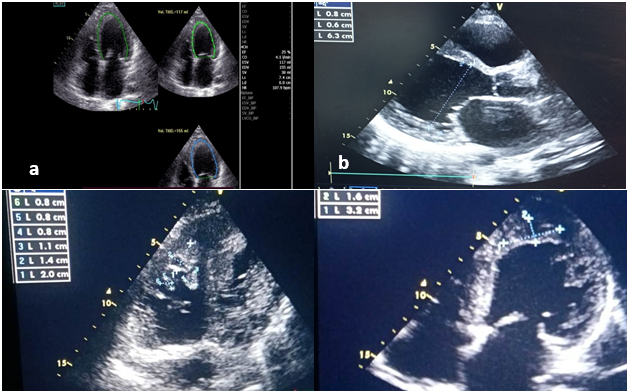
Figure 1: Transthoracic echography 2D initial evaluation: a) Severe left ventricle dysfunction by automatical systolic ejection fraction; b) large dilatation of left ventricle; c) Magma of thrombus in right ventricule apex; d) adherent thrombus in left ventricle apex.
Transthoracic echocardiography revealed an appearance of global hypokinetic heart disease in the dilated stage (TDD/TSD: 63/55 mm), with severe systolic dysfunction at 25% (SBP), low cardiac output at 1.7 l/min with the presence of an adherent left apical thrombus measuring 21 x 40 mm, and several right apical thrombi, the largest of which measured 12 x 23 mm. There was no significant mitro-aortic valve disease. The mitral profile was restrictive, indicating increased left ventricular filling pressures. The atria were dilated; the right ventricle was undilated with moderate longitudinal systolic dysfunction. There was pulmonary hypertension at 60 mmHg with a dilated inferior vena cava (figure 2).
The patient was depleted by injectable furosemide with rapid oral relay, heart failure treatment was introduced gradually after passing the acute phase. It was decided to start SGLT2 inhibitors but their high cost was a barrier to the patient. Oral anticoagulation with vitamin K antagonists was initiated with a target INR of 2-3. After discussion with the internists, the patient was given a bolus of corticosteroids with an oral relay. Three months later, the patient was mildly symptomatic with NYHA stage II dyspnoea, with no clinical signs of heart failure; cardiac ultrasound showed slight improvement with an ejection fraction of 35%, disappearance of the left apical thrombus, and regression of the thrombotic magma in the right ventricle (Figure 2) The global longitudinal strain was modified to -4.2% (Figure 2). We titrated the heart failure treatment progressively to an optimal dose.
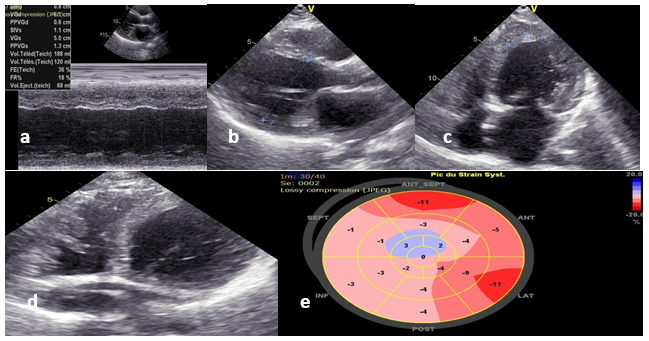
Figure 2: Control 2D TTE after 3 months: (a) and (b) Persistence of left ventricle dilatation; c) and d) Large regression of ventricular thrombus; e) Severe alteration of global longitudinal strain.
Result and Discussion
Behcet's disease is a systemic inflammatory disease of unknown etiology. Its pathophysiology is based on local and systemic occlusive vasculopathy [1,2,6-8]. Both sexes are concerned and the predominance varies according to the regions and the series. It most often occurs in young people between the ages of 30 and 40. It occurs most often in Mediterranean regions, the countries of the Middle and Far East with an estimated prevalence of 80-420 cases per 100,000 inhabitants in Turkey, on the other hand it is around 20 cases per 100,000 inhabitants; Its prevalence is lower in Europe and America, in sub-Saharan Africa, it predominates in North Africa but there are no national registers, there are only very few cases described in sub-Saharan Africa [1,2]. The role of genetic factors is incriminated, in particular the presence of the type I histocompatibility complex (HLA-B51) as well as environmental factors that are still poorly understood to this day [1,5,6]. It is a complex nosological entity manifested by mucocutaneous, ocular, joint, neurological, vascular, venous and arterial as well as cardiac damage [2,6,7,9]. Vascular involvement is one of the most frequently encountered elements in BD and is not seen in all patients; all vessels, of any caliber and at all sites can be affected: this is why BD is classified in the variable vasculitis group by the 2012 Chapel Hill International Consensus Conference [2]. Cardiac manifestations are very rare, and represent only 0.6% in an Iranian series [2], but 6% with 3,1 % of intracardiac thrombi in a Tunisian series [3]. Cardiac manifestations include myocarditis, pericarditis, endocarditis with sometimes severe valvular involvement, intracardiac thrombus, myocardial infarction, endomyocardial fibrosis and cardiac aneurysm [2,5,7-10].
Some cases of cardiomyopathy have also been described [2,6,7,10]. The diagnosis of BD is essentially based on clinical criteria because there is no reliable biological test and it associates the presence of bipolar aphthosis with skin lesions (erythema nodosum, pseudo-folliculitis, pustules, acneiform nodules) with ocular lesions (Anterior uveitis, posterior uveitis, cellular infiltrate in the vitreous body, retinal vasculitis) to a positive pathergic test within 24-48 h [1,11]. Recently, new international classification criteria assigning points to each of the manifestations were published in 2013 and include: oral aphthosis (2 points), genital aphthosis (2), ophthalmological involvement (2), cutaneous (1), central neurological (1), vascular (1), pathergic test (1). A patient with a score of at least 4 is classified as having Behcet's disease with a sensitivity of 94.8% and a specificity of 90.5% [12]. Our patient had a clinical score of 6 points (oral and genital aphthosis, vascular manifestations and positive pathergy test). She also presented biventricular large thrombi located on ventricular apex. She had bilateral distal embolism due to presence of multiple thrombus on apical site of right ventricular complicated with pulmonary infarction which infected. Our patient had dilated cardiomyopathy with reduced systolic function. The presence of dilated cardiomyopathy in MB is very rare, it is either related to an ischemic or inflammatory component; inflammatory damage is explained by the action of pro-inflammatory cytokines on left ventricular function. Clinically, it is manifested by systolic or diastolic heart failure, sometimes it is asymptomatic systolic or diastolic dysfunction [2,4,6,7]. The treatment is based on the administration of colchicine, immunosuppressor; corticosteroid therapy is the cornerstone of treatment for Behcet's disease flares [1,2,8]; in the event of thrombosis, treatment is based on the initiation of anticoagulant treatment [1,2,8,10-13]. Our patient was put on oral anticoagulant, heart failure treatment indicated by European society of cardiology. The corticotherapy was initiated. The prognosis depends on the presence or absence of complications, it is often favorable under treatment and seems to be more severe in young subjects [1,10,12-14]. The prognosis of our patient will depend on several parameters including adherence to treatment, response to corticoids and immunosuppressive therapy on the one hand and on the other the possibility of thrombi migration may make the patient's evolution difficult. Left ventricular dysfunction is a predictive factor for rhythmic disorders. After 3 months of treatment, we found that the patient gained some systolic function but the overall strain remained impaired; the regression of thrombi on antivitamin K was clear. Recovery of systolic function is often slow and will also depend on the degree of myocardial damage. This highlights the value of cardiac MRI to assess the degree of myocardial damage but we did not have access to this examination.
Conclusion
Behcet's disease is a multisystemic vasculitis of unknown etiology that occurs in countries along the ancient Silk Road. It affects the young subject between 30 and 40 years old, with a male predominance. Cardiovascular damage is rare but makes the prognosis pejorative. The diagnosis is clinical and is based on a clinical score. The treatment is based on anti-inflammatories and immunosuppressants, corticosteroid therapy is the cornerstone of the treatment of flare-ups, anticoagulation is required in the event of venous or arterial thrombosis, in the event of heart failure the treatment must follow the recommendations of the learned societies. The prognosis depends on the severity of each manifestation.
References
1. Bettiol A, Alibaz-Oner F, Direskeneli H, Hatemi G, Saadoun D, Seyahi E, Prisco D, Emmi G. Vascular Behçet syndrome: from pathogenesis to treatment. Nature Reviews Rheumatology. 2023 Feb;19(2):111-26. https://doi.org/10.1038/s41584-022-00880-7
2. Demirelli S, Degirmenci H, Inci S, Arisoy A. Cardiac manifestations in Behcet's disease. Intractable & rare diseases research. 2015;4(2):70-5. https://doi.org/10.5582/irdr.2015.01007
3. Kechida M, Salah S, Kahloun R, Klii R, Hammami S, Khochtali I. Cardiac and vascular complications of Behçet disease in the Tunisian context: clinical characteristics and predictive factors. Advances in Rheumatology. 2019 Jul 29;58:32. https://doi.org/10.1186/s42358-018-0032-x
4. Akdeniz N, Elmas ÖF, Karada? AS. Behçet syndrome: a great imitator. Clinics in Dermatology. 2019 May 1;37(3):227-39. https://doi.org/10.1016/j.clindermatol.2019.01.001
5. Z Tazi Mezalek, H Khibri, S El Fari, S Chadli, W Ammouri and al. Vascular manifestations of Behcet’s disease. La Revue de Médecine Interne 2023;44(2):72-8. https://doi.org/10.1016/j.revmed.2022.11.011
6. Zeidan MJ, Saadoun D, Garrido M, Klatzmann D, Six A, Cacoub P. Behçet’s disease physiopathology: a contemporary review. Autoimmunity Highlights. 2016 Dec;7(4):1-2. https://doi.org/10.1007/s13317-016-0074-1
7. Hatemi G, Seyahi E, Fresko I, Talarico R, Hamuryudan V. One year in review 2018: Behçet’s syndrome. Clin Exp Rheumatol. 2018 Jan 1;36(Suppl 115):S13-27.
8. Farouk H, Zayed HS, El-Chilali K. Cardiac findings in patients with Behcet’s disease: facts and controversies. Anatolian journal of cardiology. 2016 Jul;16(7):529-33. https://doi.org/10.14744/AnatolJCardiol.2016.7029
9. Davatchi F, Chams-Davatchi C, Shams H, Shahram F, Nadji A, Akhlaghi M, Faezi T, Ghodsi Z, Sadeghi Abdollahi B, Ashofteh F, Mohtasham N. Behcet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert review of clinical immunology. 2017 Jan 2;13(1):57-65. https://doi.org/10.1080/1744666X.2016.1205486
10. Y?ld?r?m R, Dinler M, Bilge N?, Ka?ifo?lu T. A rarely seen manifestation in Behcet’s disease: intracardiac thrombosis. Clinical Rheumatology. 2021 Oct;40(10):4355-6. https://doi.org/10.1007/s10067-021-05720-9
11. Desbois AC, Terrada C, Cacoub P, Bodaghi B, Saadoun D. The ocular manifestations of Behçet's disease. The Journal of Internal Medicine. 2018 Sep 1;39(9):738-45. https://doi.org/10.1016/j.revmed.2018.02.022
12. L Guillevin. Le livre de l’interne: Médecine interne 2ème édition. Médecine sciences Publications Lavoisier Paris 2014;48:407-15. https://complements.lavoisier.net/9782257205032_medecine-interne-2-ed-coll-le-livre-de-l-interne_Sommaire.pdf
13. Mehdipoor G, Davatchi F, Ghoreishian H, Shabestari AA. Imaging manifestations of Behcet’s disease: key considerations and major features. European journal of radiology. 2018 Jan 1;98:214-25. https://doi.org/10.1016/j.ejrad.2017.11.012
14. Ouedraogo SM, Djibril MA, Mossi E, Balaka A, Tchamdja T, Djagadou A, Ilboudo A, Bagbila A, Kaaga L, MOukaila A. Maladie de Behçet: A propos d’un cas au CHU Sylvanus Olympio de Lomé et revue de la littérature. Revue Africaine de Médecine Interne. 2017 Jun 19;4(1-1):61-5.